Getting started with network plots (Last updated: 2025-06-03)
The visNetwork package greatly simplifies the process of producing useful, interactive network plots.
I’ve long been hoping for a reason to have to devote time to learning how to produce network plots. In my world, where bar and line charts reign supreme (with heatmaps and waffle charts thrown in occasionally) it is nice to be able to develop a new visualisation.
I’ve been wanting to produce a network plot for some time. But, the data structure, with its nodes and edges, and seeming lack of any identifiable characteristics, has meant it has not been hugely far up my agenda, or at least, never far up enough to make me learn more about it.
The TL/DR on this was that I was asked to join up some tables that held details of patients who had tested positive for Covid-19, and their known contacts. The suspicion was that there might be cases who were both an initial index case (tested positive) and a contact for another case. Colleagues in other depts could easily get the dataset and filter it to their hearts content in Excel to try and figure this out.
Others could write endless SQL queries, but I surmised that a network plot would tell us everything we needed to know, at once.
Lets think of a scenario using Bob, Bill and Brenda as our persons of interest (there is nothing significant about the names)
For example, Bill tests positive, but was unaware of this when he met Brenda a few days earlier. It turns out that prior to that, Bill also met up with Bob, who not only bought him a coffee, but passed on the virus.
In this scenario Bob is an index case, Brenda is a contact, and Bill is both.
I generated some mock ID data using Mockaroo.
In Scotland, we have CHI (Community Health Index) numbers (which are 10 digits long). In England and Wales there are NHS Numbers In all cases these are unique identifiers to designate patients.
In my example data, Mockaroo generated mock SSN numbers. It doesn’t matter, the main point is I have (made up) unique IDs for each patient.
I’ve created a small repo with the dummy data and code, so if you want to follow along, head over to the repo on github
Let’s read them in..
library(data.table) # note cases will be both a data.table and a data.frame library(dplyr) library(tibble) library(tidyr) library(visNetwork) cases <- data.table::fread("MOCK_DATA.csv") %>% filter(complete.cases(.)) It is very important to ensure you have no missing values, otherwise NULL / NA’s will link to each other and produce a very distorted view of the data.
Distinct index cases and contacts
We have two columns, id1 and id2. id1 are our index cases - the positive patients. id2 are our contacts - people who have been in contact, but who may or may not have the virus themselves.
First, we obtain unique values for both columns, and rename the resulting colum to ‘label’:
sources <- cases %>% distinct(id1) %>% rename(label = id1) contacts <- cases %>% distinct(id2) %>% rename(label = id2) Then we join them together to get a final tibble of ids, and provide a new unique id for each row. These create the nodes for our network.
Nodes is just a fancy term for a data point.
nodes <- full_join(sources, contacts, by = c("label")) nodes <- nodes %>% rowid_to_column("id") head(nodes) ## id label ## 1: 1 536-10-8682 ## 2: 2 280-30-2349 ## 3: 3 214-05-0872 ## 4: 4 712-65-9980 ## 5: 5 356-34-3345 ## 6: 6 164-38-1022 Now we need to figure out how many different combinations we have. In this case, we probably don’t expect to have more than 1 instance per combination, but for other use cases, you might need to know how many times these combinations appear, so we will leave the ‘weight’ calculation in.
per_case <- cases %>% group_by(id1, id2) %>% summarise(weight = n()) %>% ungroup() ## `summarise()` regrouping output by 'id1' (override with `.groups` argument) Again, this time round, there is no difference between leaving summarise as is, or providing ‘keep’ to the .groups argument.
head(per_case,10) ## # A tibble: 10 x 3 ## id1 id2 weight ## <chr> <chr> <int> ## 1 164-38-1022 177-96-6142 1 ## 2 164-38-1022 295-98-8195 1 ## 3 164-38-1022 801-50-4484 1 ## 4 180-90-3238 407-90-2819 1 ## 5 180-90-3238 595-05-8736 1 ## 6 188-07-5552 620-43-5564 1 ## 7 201-94-4265 189-87-2804 1 ## 8 201-94-4265 536-10-8682 1 ## 9 201-94-4265 558-62-5961 1 ## 10 214-05-0872 412-54-2941 1 We now turn our attention to the edges - the connecting lines between the points (nodes).
We join our per_case tibble to the nodes, by the id1 value, so that we return the rownumber (id) of the matching node. We rename this as our ‘from’ column
edges <- per_case %>% left_join(nodes, by = c("id1" = "label")) %>% rename(from = id) head(edges,5) ## # A tibble: 5 x 4 ## id1 id2 weight from ## <chr> <chr> <int> <int> ## 1 164-38-1022 177-96-6142 1 6 ## 2 164-38-1022 295-98-8195 1 6 ## 3 164-38-1022 801-50-4484 1 6 ## 4 180-90-3238 407-90-2819 1 15 ## 5 180-90-3238 595-05-8736 1 15 We do the same by matching id2, and returning a ‘to’ value
edges <- edges %>% left_join(nodes, by = c("id2" = "label")) %>% rename(to = id) head(edges,5) ## # A tibble: 5 x 5 ## id1 id2 weight from to ## <chr> <chr> <int> <int> <int> ## 1 164-38-1022 177-96-6142 1 6 41 ## 2 164-38-1022 295-98-8195 1 6 45 ## 3 164-38-1022 801-50-4484 1 6 25 ## 4 180-90-3238 407-90-2819 1 15 47 ## 5 180-90-3238 595-05-8736 1 15 34 We can verify this - let’s check the label for the node with an id of 6:
nodes[id == 6,] ## id label ## 1: 6 164-38-1022 So, to be clear, the ‘from’ and ‘to’ are simply returning the row id numbers for the corresponding unique patient identifier. We don’t need the patient identifiers themselves for these connecting lines
edges <- select(edges, from, to, weight) head(edges,5) ## # A tibble: 5 x 3 ## from to weight ## <int> <int> <int> ## 1 6 41 1 ## 2 6 45 1 ## 3 6 25 1 ## 4 15 47 1 ## 5 15 34 1 Are there any more Bills?
Now we want to know if there is anyone who is both an index and a contact. The plan:
- Return the labels from sources and contacts as separate vectors
- Find the index positions of any patients from sources who are also in contacts
- Create a new column to identify the sources as index cases
To do this, we turn the ‘sources’ vector to a tibble using as_tibble_col, then mutate a new descriptor column.
Because we read in the data originally using data.table’s fread, sources[related,] constructs a data.table, so we do not need to use as_tibble_col again.
Now we have 2 tibbles/data.frames/data.tables that have a ‘label’ and ‘record_type’ column.
sourcesvec <- sources$label contactsvec <- contacts$label related <- which(sourcesvec %in% contactsvec) sources_id <- sourcesvec %>% as_tibble_col(.,column_name = 'label') %>% mutate(record_type = "index") related_id <- sources[related,] %>% # as_tibble_col(., column_name = 'label') %>% ## I don't need this row because I return a data.table/ data.frame above) mutate(record_type = "index_and_contact") We bind these last two tables together to form a lookup for the nodes table:
index_lookup <- bind_rows(sources_id,related_id) Then we do the join:
nodes <- nodes %>% left_join(index_lookup, by = "label") head(nodes, 5) ## id label record_type ## 1: 1 536-10-8682 index ## 2: 1 536-10-8682 index_and_contact ## 3: 2 280-30-2349 index ## 4: 3 214-05-0872 index ## 5: 4 712-65-9980 index Some of the record_types are contacts, and are now ‘NA’.
They should be ‘contact_only’, so tidyr to the rescue, specifically, replace_na:
nodes <- nodes %>% mutate(record_type = replace_na(record_type,"contact_only")) head(nodes, 5) ## id label record_type ## 1: 1 536-10-8682 index ## 2: 1 536-10-8682 index_and_contact ## 3: 2 280-30-2349 index ## 4: 3 214-05-0872 index ## 5: 4 712-65-9980 index nodes$shadow <- TRUE # Nodes will have a drop shadow nodes$title <- nodes$label # Text on click nodes$label <- nodes$label # Node label We can also now add borders, and determine how things are higlighted when clicked.
nodes$color.border <- "black" nodes$color.highlight.background <- "orange" nodes$color.highlight.border <- "darkred" We are getting there, trust me. In my real life case, I had a couple of thousand points. Some of those were both index, and a contact, and so were appearing more than once in my nodes list.
If they were an index_and_contact, I wanted the plot to reflect that.
So to be doubly sure, I wanted to find any times where an id appeared more than once, and remove any rows where the record_type was ‘index’.
The plan:
- First, find the duplicates
- Create a new table from their values
- Anti_join them with the nodes table to remove them
#some nodes are both index, and index and contact # we need to dedupe them, so we get rid of the index dupes <- nodes %>% group_by(id) %>% tally() %>% filter(n >= 2) %>% pull(id) nodes_to_remove <- nodes %>% filter(id %in% dupes & record_type == 'index') nodes <- anti_join(nodes, nodes_to_remove) ## Joining, by = c("id", "label", "record_type", "shadow", "title", "color.border", "color.highlight.background", "color.highlight.border") We can format the edges (connecting lines)
edges$color <- "gray" # line color edges$arrows <- "middle" # arrows: 'from', 'to', or 'middle' edges$smooth <- FALSE # should the edges be curved? edges$shadow <- FALSE # edge shadow Finally, in order to take advantage of visNetwork’s capabilities, we’ll rename the record_type column to ‘group’
nodes <- nodes %>% rename(group = record_type) head(nodes, 5) ## id label group shadow title color.border ## 1: 1 536-10-8682 index_and_contact TRUE 536-10-8682 black ## 2: 2 280-30-2349 index TRUE 280-30-2349 black ## 3: 3 214-05-0872 index TRUE 214-05-0872 black ## 4: 4 712-65-9980 index TRUE 712-65-9980 black ## 5: 5 356-34-3345 index TRUE 356-34-3345 black ## color.highlight.background color.highlight.border ## 1: orange darkred ## 2: orange darkred ## 3: orange darkred ## 4: orange darkred ## 5: orange darkred Why? So we can pass in formatting such as colour and shape, based on the ‘groupname’ property. Here, based on the group ( or record_type as it was previously) we set index values to be firebrick red with a diamond shape, and so on. The last line adds a legend. Unfortunately, this can only be to the left or right, and not at the bottom of a plot
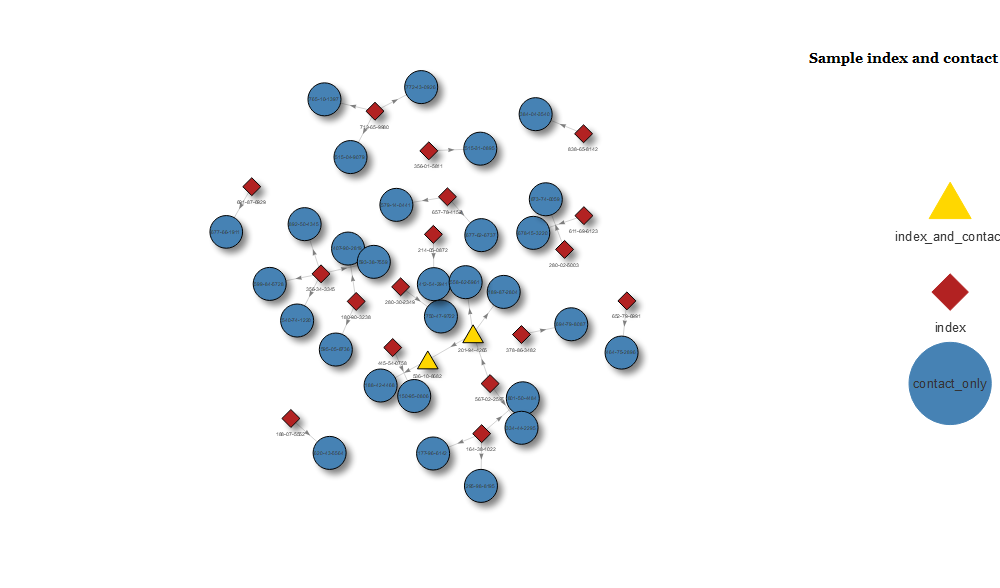
visnet <- visNetwork(nodes, edges) %>% visGroups(groupname = "index", color = "firebrick", shape = "diamond") %>% visGroups(groupname = "index_and_contact", color = "gold", shape = "triangle") %>% visGroups(groupname = "contact_only", color = "steelblue", shape = "circle") %>% visLegend(position = "right", main = "Sample index and contact") visnet
The resulting plot is fully interactive. You can scroll in, click on points, drag them around etc. It’s marvellous, and it really impressed those who I showed it to.
There is no way I could create this with some of the more traditional BI tools that are currently being used in my organisation. Yet another win for R!